

Written by Julie Bick, Ph.D.
A companion diagnostic (CDx) is a term used to describe an in vitro assay using advanced molecular or omics technologies that are developed alongside a specific drug to identify patients who are most likely to benefit from that drug. This concept of CDx has been the cornerstone of Precision Medicine- an approach that leverages a patient’s genetic and phenotypic profile to develop tailored therapeutic strategies with maximum safety and efficacy, and minimum risk of adverse reactions or side effects.
The adoption of CDx into the drug development industry has been rapid and transformative, from accelerating drugs through the approval process and identifying responding cohorts of patients more rapidly, to reducing the costs of treatment across all industry stakeholders.
The approach taken for the selection of a biomarker for a CDx test is driven mainly by its overall role. Is this biomarker predictive of drug efficacy, or used for pharmacodynamic, prognostic, or safety profiling of patients? The identification of the most appropriate and fit-for-purpose biomarker is central to precision medicine approaches and there is certainly no one-size-fits-all. Like many aspects of healthcare, the CDx approach is evolving to overcome the limitations of a one drug/one biomarker approach. In this blog, we explore the successes and limitations of CDx approaches and the new paradigm shift in the development of Next Generation CDx tests (NGCDx).
The development of a first-generation companion diagnostic test involves several phases during which the biomarker’s significance increases under the auspices of a clinical trial. This is accompanied by changes in the regulatory landscape of the workflow which dictates how the tests are performed and the data managed (Fig. 1).
The first step in developing a CDx is identifying a biomarker that can be used to predict response to the drug. In some instances, the selection of the biomarker is obvious since it is the target of the drug itself- this is often the case for oncology drugs that target specific proteins driving tumor growth. In other instances, identifying a biomarker is more complex and requires screening patient samples or using data from pre-clinical studies to identify potential biomarker candidates.
Once a potential biomarker has been identified, it must be validated in a larger cohort of patients. This can be quite a complex process, and different technologies may need to be assessed to determine which assay endpoints provide the most correlative data. Although immunohistochemistry (IHC) for protein biomarker measurement can be accurate, it may not be a scalable platform for larger drug trials, whereas RNase Seq may not provide a reliable readout of the expressed levels of protein biomarkers. This whole process usually involves analyzing patient samples from clinical trials to determine whether the selected biomarker is truly predictive of treatment response and patient outcome.
Once the biomarker has been validated and shown to be highly correlative with the drug target, a diagnostic test must be developed to detect it. This typically involves developing an assay that can detect the presence and level of the biomarker in blood or tissue samples.
The diagnostic test must then be validated to ensure that it is accurate and reliable within the clinical trial environment. This may involve analyzing many patient samples to assess the sensitivity and specificity of the test and validate it in accordance with CLIA and CAP guidelines.
If the CDx is run as a Laboratory Derived Test (LDT), then it can only be performed in the CLIA laboratory that has performed the validation. Although this can work for some CDx applications, the next step for most advanced CDx assays is the subsequent approval by regulatory agencies such as the FDA or EMA enabling the test to be run in multiple laboratories. The regulatory process may involve submitting data from clinical trials or analytical studies to demonstrate the safety and efficacy of the test. This can be a time-consuming and expensive undertaking and usually involves a partnership with a specialty clinical test company.
Finally, the CDx must be commercialized and made available to patients. This may involve partnering with a diagnostic company to manufacture and distribute the test, or integrating the test into the drug development process so that it is available to physicians at the time of treatment.
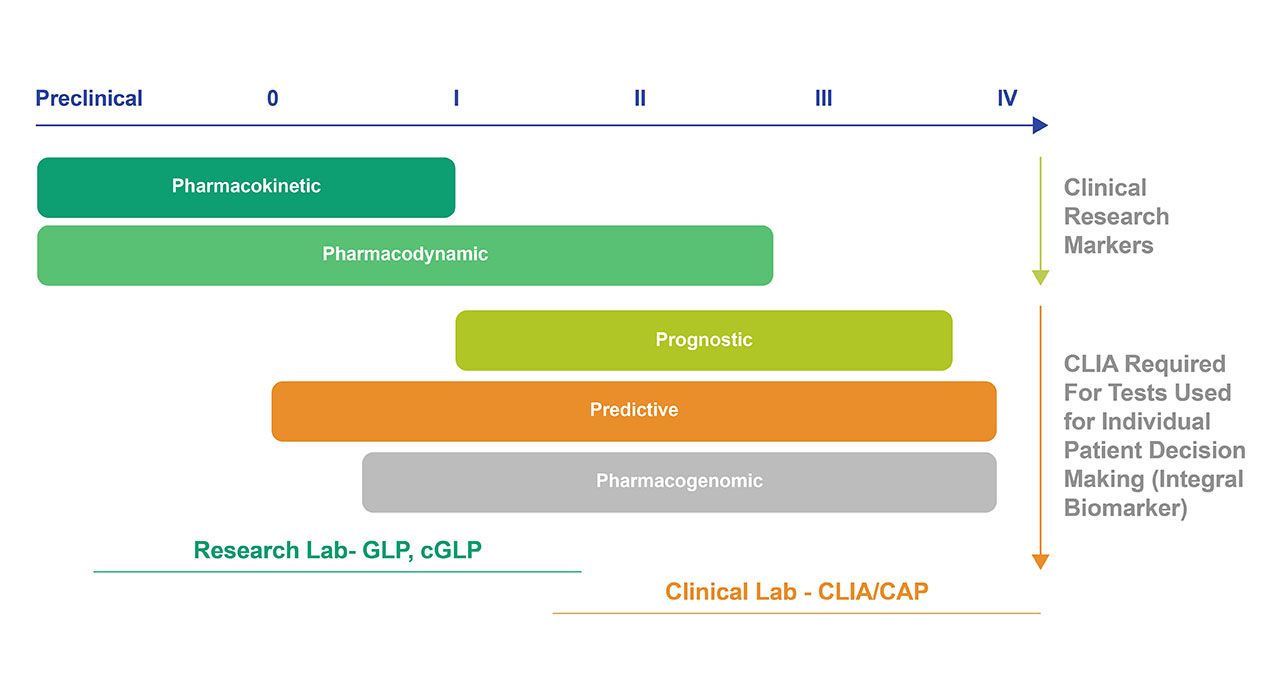
Fig 1. Classes of Biomarkers throughout the Drug Development Process. Integrated Markers are used in research studies and not for medical decision making, but rather to test a hypothesis. These types of biomarkers can be analyzed in non-clinical laboratories in the format of a CDx. Once the clinical evaluation has progressed to Phase II and beyond, the biomarkers are considered Integral Markers – these can be used for medical decision making, and results are reported to the patients or physicians- these CDx tests must be performed in a CLIA-approved laboratory.
One of the earliest and most successful CDx approaches was for HER2 testing for breast cancer. HER2 is a protein that is overexpressed in some types of breast cancer, and the testing of HER2 status has helped identify patients who are most likely to benefit from treatment with targeted therapies such as trastuzumab.
The companion diagnostic test for HER2 is typically performed using a tissue sample obtained from a biopsy of the tumor. The sample is then analyzed using one of several techniques, including IHC and/or fluorescence in situ hybridization (FISH).
IHC uses anti-HER2 antibodies to stain the tissue and label any HER-2 protein. The level of staining is then evaluated under a microscope and given a score between 0 and 3+. A score of 0 or 1+ indicates that the tumor is HER2 negative, while a score of 2+ requires further testing using FISH. A score of 3+ indicates that the tumor is HER2-positive. FISH is a semi-quantitative method using fluorescent probes to bind to the HER2 gene and detect its copy number in the tumor cells. The ratio of HER2 gene copies to chromosome 17 copies is then calculated, and a ratio greater than 2.0 indicates that the tumor is HER2-positive.
Both IHC and FISH are widely used in clinical practice to determine HER2 status, and both tests have high levels of sensitivity and specificity. However, they require specialized laboratory expertise and equipment to perform accurately. Therefore, they are typically performed by specialized pathology laboratories rather than general clinical laboratories.
Although certainly a significant step forward in truly personalized therapeutic approaches, traditional CDx tests were developed along a linear concept of one drug- one biomarker. This turned out to be overly simplistic and limited in the test’s ability to support safe and effective therapeutic strategies. This was particularly significant when addressing solid tumors that were diagnosed with limited tissue specimens. In essence, there are recognized heterogeneity within any cancer type, and between patients with the same type of cancer- these differences in genetic, epigenetic, and phenotypic backdrops can profoundly influence the outcome of therapeutic intervention (Deng, N. et. al 2012). In an open letter, Kurzrock and his team (Kurzrock, R. et. al. 2014) called for a comprehensive overhaul in the regulation of clinical testing, and many joined in supporting the adoption of multiplexed analysis of biomarkers to overcome the limitations imposed by cancer heterogeneity (Khoury and Catenacci, 2014). This was the catalyst for the foundation and widespread adoption of NGCDx testing methods.
At their core, NGCDx approaches combine cutting-edge techniques including genomics, proteomics, and metabolomics to analyze a patient's biomarkers to identify specific genetic mutations or other molecular abnormalities that may be driving the patient's disease. The concept was to use these data points to help clinicians tailor a series of treatments to individual patients, and therefore increase the likelihood of successful outcomes and reduce the risk of side effects. In addition, the rich datasets generated using these tests can help researchers identify new drug targets, understand disease mechanisms, drug mechanisms of action, and assess the safety and efficacy of new therapies.
Common NGCDx approaches include NGS genome sequencing, and mass spectroscopy proteomic profiling of patients, to better understand the genotypic and phenotypic signatures of their cancer. The first FDA-approved NGCDx was FoundationOne CDx for ovarian cancer and has been followed by several others.
EGFR testing for lung cancer: Epidermal growth factor receptor (EGFR) mutations are found in a subset of lung cancers. Testing for EGFR mutations can help to identify patients who are most likely to benefit from treatment with EGFR inhibitors.
The NGCDx test for EGFR mutations is typically performed using a tissue sample obtained from a biopsy of the tumor, using one of several techniques, including PCR (polymerase chain reaction), Sanger sequencing, and next-generation sequencing (NGS). PCR and Sanger sequencing are both used to detect EGFR mutations in tumor samples. NGS is a more recent and advanced technique that can detect multiple mutations in multiple genes simultaneously. It involves sequencing the entire DNA of the tumor sample and comparing it to a reference genome to identify any mutations.
All three techniques have high levels of sensitivity and specificity and can accurately detect EGFR mutations. However, NGS has the advantage of detecting multiple mutations simultaneously and can therefore be useful in identifying patients who may be candidates for targeted therapies beyond EGFR inhibitors. (Douillard JY, et al. 2013).
PD-L1 testing for cancer immunotherapy: PD-L1 is a protein that is expressed in some types of cancer cells. Testing for PD-L1 status can help to identify patients who are most likely to respond to immunotherapy drugs such as pembrolizumab and nivolumab. The NGCDx test for PD-L1 expression is typically performed using a tissue sample obtained from a biopsy of the tumor that is assessed using IHC to determine the level of PD-L1, and a score given based on the percentage of tumor cells that express PD-L1 on the cell surface. The scoring systems used by different tests may vary, but they generally classify PD-L1 expression into several levels, such as negative, low, intermediate, and high.
The test used to detect PD-L1 expression may vary depending on the specific therapy being used. Different PD-L1 inhibitors may have different requirements for PD-L1 expression levels to be effective. Therefore, the choice of test should be guided by the clinical context and the specific needs of the patient. It is important to note that PD-L1 expression can be dynamic and may change over time, especially in response to therapy. Therefore, repeat testing may be necessary to determine if a patient is still eligible for PD-L1 inhibitor therapy or if their treatment needs to be modified.
BCR-ABL testing for chronic myeloid leukemia: BCR-ABL is a fusion protein that is found in most cases of chronic myeloid leukemia (CML). Testing for BCR-ABL can help to monitor response to treatment with tyrosine kinase inhibitors. The test for BCR-ABL is typically performed using a blood or bone marrow sample obtained from the patient. The sample is then analyzed using either reverse transcriptase-polymerase chain reaction (RT-PCR) to detect BCR-ABL transcripts, or FISH to detect the presence of the BCR-ABL fusion gene. RT-PCR can detect even very low levels of BCR-ABL transcripts and is used to monitor disease progression and response to therapy in patients with chronic myeloid leukemia (CML). FISH is particularly useful in cases where the BCR-ABL fusion gene is present in only a small proportion of cells, or where the sample quality is not adequate for RT-PCR analysis.
Both RT-PCR and FISH are highly sensitive and specific methods for detecting the BCR-ABL fusion gene, and they are widely used in clinical practice to diagnose and monitor patients with CML. The choice of the test may depend on the specific needs of the patient and the laboratory performing the analysis.
KRAS testing for colorectal cancer: KRAS mutations are found in a subset of colorectal cancers. Testing for KRAS mutations can help to identify patients who are unlikely to respond to treatment with EGFR inhibitors. The NGCDx test for KRAS mutations is typically performed using a tissue sample obtained from a biopsy of the tumor. The sample is then analyzed using one of several techniques, including PCR, Sanger sequencing, and NGS.
Whereas PCR is a widely used method for detecting KRAS mutations in tumor samples, NGS can detect multiple mutations in multiple genes simultaneously through the sequencing of the entire DNA of the tumor sample and comparing it to a reference genome to identify any mutations.
All three techniques have high levels of sensitivity and specificity and can accurately detect KRAS mutations. However, NGS has the advantage of detecting multiple mutations simultaneously and can therefore be useful in identifying patients who may be candidates for targeted therapies beyond KRAS inhibitors.
The adoption of NGCDx protocols into clinical trials has invoked several different approaches to stratifying patient cohorts within trials to best understand the role biomarkers play and expedite the expansion of approved drugs into off-label therapeutic strategies (Khoury JD, Catenacci DV., 2015). Here are two examples of modified clinical trial strategies that incorporate advanced NGCDx Testing methods.
The Expansion Platform Type II clinical trial design allows for the testing of multiple treatments in a single trial. The goal is to evaluate the effectiveness of multiple treatments for a specific disease or condition while reducing the overall time and cost of conducting multiple separate trials. The Expansion Platform Type II Design trial is divided into two stages. In the first stage, a smaller sample size is used to test the primary endpoint of the study. After the interim analysis of the data from the first stage, the trial sponsor can decide whether to stop the study early, continue the study with the original sample size, or expand the sample size to test secondary endpoints or other outcomes of interest.
The inclusion criteria for patients are based on multiple biomarkers, not just the drug target determined through omics analysis. The trial includes multiple treatment arms, each testing a different treatment or combination of treatments, and each arm is evaluated independently for its effectiveness. It includes a control arm, which typically receives the standard of care treatment or a placebo. As data is collected from each treatment arm, additional patients may be enrolled into the treatment arm showing the most promise. Statistical methods are then used to compare the effectiveness of each treatment arm with the control arm and to evaluate the overall efficacy of the treatments being tested.
This approach has several advantages over traditional trial designs, including the ability to test multiple treatments simultaneously and the flexibility to adjust treatment arms based on the results of the trial. If the trial is expanded, additional patients are enrolled in the second stage, and the data from the two stages are combined to analyze the primary and secondary endpoints of the study.
The Expansion Platform Type II Design is particularly useful for clinical trials in areas where there is a high degree of uncertainty, such as in rare diseases, where the sample size may be limited, or in areas where new treatments are being tested, and the effect size is unknown. By allowing for adaptive modifications to the sample size based on interim data, the design can help to optimize the study's statistical power and efficiency, while reducing the risk of false-positive or false-negative results (Tesfaye et. al. 2019).
The BATTLE (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination) clinical trial process for lung cancer involves several stages of testing and evaluation to determine the safety and efficacy of a new treatment approach for lung cancer patients (Papadimitrakopoulou V., et. al. 2016).
The BATTLE clinical trial process is unique in several ways; firstly, the trial uses a comprehensive biomarker-based approach to identify the best treatment options for individual patients by analyzing for genetic mutations and other biomarkers in each patient's tumor tissue. Additionally, the trial employs an adaptive design, meaning that treatment strategies can be adjusted based on the results of earlier treatments. This allows for a more personalized approach to treatment and allows clinicians to identify which treatments are most effective for different patient subgroups. Within the trial, structure are various multiple treatment arms, each testing a different targeted therapy. This allows for the simultaneous evaluation of multiple treatments, reducing the time and cost required to test each treatment separately.
This is very much a patient-centered approach since the BATTLE trial is focused on improving outcomes for individual patients, rather than on developing new drugs or therapies and helps to ensure that patients receive the best possible care and that treatments are tailored to their specific needs.
The heavy reliance on the use of biomarker-based approaches, adaptive trial design, multiple treatment arms, and institutional collaboration, all coupled with a patient-centered approach have made the BATTLE trial an important model for personalized medicine and have helped to accelerate the development of more effective treatments for lung cancer patients.
Where CDx testing transformed personalized medicine and drug development, NGCDx tests are now revolutionizing clinical trials by enabling more precise patient stratification and enrollment and driving the ability to screen highly potent combination therapies more effectively. By using NGCDx to identify patients with specific biomarker combinations, clinical trial sponsors can design studies with more homogeneous patient cohorts, which improves the statistical power and reduce the variability of the results, and ultimately improve the efficiency and success rate of therapeutic strategies.
NGCDx tests are also playing a critical role in drug development by providing real-time feedback on the performance of investigational therapies. These tests can help accelerate the drug development process and bring new treatments to patients more quickly while managing costs more effectively. The paradigm shift from one biomarker-one drug was only made possible by the development of enhanced omics platforms. We can now get an incredible amount of data from clinical samples and identify genetic variants within tumors. All of this ultimately results in more precise and personalized treatment decisions, better outcomes, and improved quality of life for patients.
